Happy MedCram Monday! Today’s MedCram Video Explanation comes from the BoardVitals USMLE Step 1 and COMLEX Level 1 banks. If you are gearing up for one of these exams, one of the topics you’ll need to be comfortable in includes Glycogen Storage Diseases (GSD). One of the BoardVitals’ questions on this topic has been found particularly tricky, so we’ve called on Dr. Roger Seheult from MedCram to break it down.
The Question: A 26-year-old male developed worsening weakness in his legs for the last five months. He noticed that running and walking was becoming increasingly difficult. He does not have any chronic diseases and has not sustained any injuries to his body. Deficiency of which of the following enzymes is most likely?
A. Glycogen synthase
B. Debranching enzyme
C. Branching enzyme
D. Acid maltase
E. Glucose-6 phosphatase
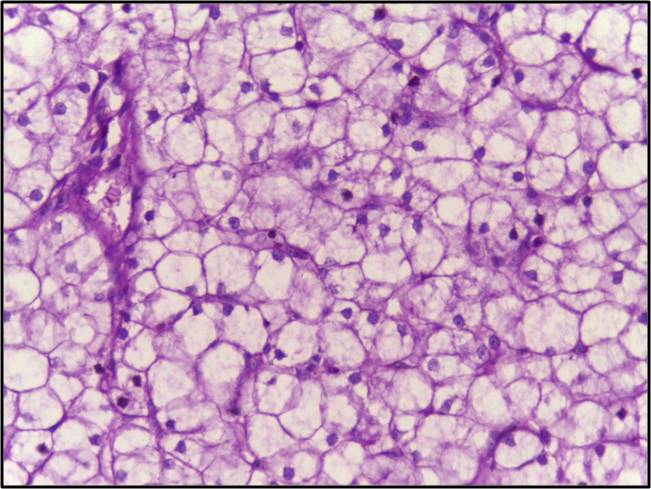
Main learning point: Pompe Disease occurs from a deficiency in acid maltase leading to accumulation of glycogen in lysosomes resulting in muscle weakness. In the infantile form patients typically die of heart failure by the age of 2 years.
Correct answer: (D) Acid maltase.
Explanation: The patient mentioned in the question most probably has a deficiency of acid maltose (Lysosomal α1→4 and α1→6 glucosidase). A deficiency of this enzyme leads to a disease known as Pompe’s (also called Glycogen storage disease type II). In the juvenile onset variant, there is an accumulation of glycogen in lysosomes and development of muscle hypotonia. Usually, the patient dies by the age of two years due to heart failure. In the adult-onset variant of this disease, there is a relative lack of cardiac involvement. The onset is more insidious and skeletal muscle involvement is more prominent especially in the lower limbs.
(B) Deficiency of the debranching enzyme leads to a disease known as Cori’s disease. There is fasting hypoglycemia and hepatomegaly in infancy. And there is an accumulation of characteristic branched polysaccharides.
(C) Deficiency of the branching enzyme leads to a disease known as Amylopectinosis or Anderson’s disease. There is hepatomegaly in infancy. And there is an accumulation of characteristic polysaccharide with few branch points. The patient usually dies in the first year of life due to heart or liver failure.
Reference: 1. David A Bender and Peter A Mayes. Metabolism of Glycogen. (Chapter 19) In: Harper’s Biochemistry. Robert K Murray, Daryl K Granner, and Victor W Rodwell. (Editors); 27th Edition. McGraw-Hill Publications; 2006
Did you get it right before watching the video? Whether you did or didn’t, practicing questions like these will help you build confidence for your upcoming exam. Check out the USMLE Step 1 or COMLEX Level 1 free trial for access to new practice questions.